












































服务电话
400-600-5039

精微高博公众号
北京精微高博仪器有限公司
Sales@jwgb.net
 比表面积分析仪
介孔孔径分析仪微孔孔径分析仪化学吸附仪反应评价装置蒸汽吸附仪穿透曲线分析仪真密度仪压汞仪高压吸附仪热分析仪X射线衍射仪脱气机质谱仪相关配件
比表面积分析仪
介孔孔径分析仪微孔孔径分析仪化学吸附仪反应评价装置蒸汽吸附仪穿透曲线分析仪真密度仪压汞仪高压吸附仪热分析仪X射线衍射仪脱气机质谱仪相关配件 化学吸附仪
蒸汽吸附仪
穿透曲线分析仪
真密度仪
压汞仪
脱气机
质谱仪
相关配件
化学吸附仪
蒸汽吸附仪
穿透曲线分析仪
真密度仪
压汞仪
脱气机
质谱仪
相关配件

Frequently Asked Question
1) 泵油加的过多,检查视窗液面高度,确保在刻度线以内,若无刻度线,确保高度不超过2/3,过多使用时有可能会溅出
2) 机械泵抽空气,抽真空管路联通空气,长时间抽空气时泵油会飞溅出来,同时机械泵会有很大噪音且冒白烟状态出现
用 CO₂ 在 273K 可以测量的最大相对压力为 P/P₀ =0.03,因此该方法只能用于研究 1 nm以下的微孔。CO₂ 在 273K 的吸附已成为研究具有极窄微孔的含碳材料的理想方法。然而,CO₂ 不能用于具有极性表面基团的微孔固体(如:氧化物,沸石,MOF 材料)的孔径分析,因为 CO₂ 的四极矩作用比 N₂ 的还大。样品要涉及到介孔大孔分析的时候,不合适用CO₂吸附数据作分析。
平均孔径:通过总孔体积及比表面积计算而来,假设样品的孔都是圆柱形孔,平均孔径=4V/A
中值孔径:吸附量达到一半或者孔体积达到一半时对应的孔径
最可几孔径:数量最丰富的孔的孔径
当最可几孔径与中值相差较小时,说明孔径分布比较集中,当最可几孔径与中值相差较大时,说明孔径分布宽泛,不同孔径数量占有较大比例;中值孔径与多点BET比表面积无相关性,不能构建关联
氢氧化钙在空气中存储会吸水分,导致内部结构发生微小改变,比表面积进而变化;即使按照同样的处理条件处理,但因为结构的变化也会导致结果的变化,建议后续测试时使用新样品,不要测试对比长时间在空气中存储的样品
此液体不太可能是水分,水分在真空加热条件下比较容易处理,多为有机物,此情况银粉等生产过程添加有机溶剂的样品容易出现;有其他颜色代表样品也挥发出水分之外的东西
此种情况基本都会对样品本身的结构造成影响,导致测试结果异常,需要降低处理温度,确保不破坏样品结构
此现象为结束实验的方式选择为降杯抽真空,结束实验后样品管内是负压,强行拆样品管时空气瞬间进入样品管,导致大量样品被吹飞,需要拆卸样品管卡头,清理烧结滤芯,严重抽飞需要做保压检漏,若存在漏气现象,需要清理管路及电磁阀
拆样品管之前必须确认样品管内压力恢复至常压,才能进行拆卸,若拆的时候感觉有吸力,需要用力才能拆下来,则代表内部负压,需要立即拧紧卡头,仪器充气至常压再拆卸样品管
若在电梯上升或者下降过程顶到或者压到东西,需要立即仪器断电,断电后电梯会停止动作,重新开机建立连接后,根据情况选择电梯上升或者下降,移走障碍物后,按照正常流程进行
若上升过程把样品管顶碎,恢复以后需要小心拆卸样品管,戴上手套以免划伤,清理碎渣
电梯升降若被顶变形,上升或者下降碰到障碍物都有可能导致变形,变形后液氮杯放置时可能会出现不稳情况,需要更换新升降
1) 若液氮杯在下方未上升,电路恢复后重连仪器,按照正常流程重新进行即可
2) 若液氮杯已经在上方,电路恢复后重连仪器,点击预抽(BK100或者BK112型号需要确保每个站都装有空样品管或者金属堵头),待压力降至0.1kPa以下,保持预抽,点击下降,液氮杯下降以后拿走放到安全位置,等待样品管恢复室温,恢复室温后,样品重新预处理,后续按照正常步骤进行
1) 若液氮杯在下方未上升,电路恢复后重连仪器,按照正常流程重新进行即可
2) 若液氮杯已经在上方,电路恢复后重连仪器,点击预抽(BK100或者BK112型号需要确保每个站都装有空样品管或者金属堵头),待压力下降至0.1kPa以下,保持预抽,点击下降,液氮杯下降以后拿走放到安全位置,等待样品管恢复室温,恢复室温后,样品重新预处理,后续按照正常步骤进行
1) 漏气导致,若突然或者偶尔出现,多为样品管安装不合适,或者密封圈及样品管有破损,密封圈和金属垫环顺序装反导致,排查解决
2) 机械泵没有打开
3) 样品一直挥发气体,达到一个类似压力动态平衡的状态,导致压力抽不下去,延长时间后压力会下降
错误操作导致泵油倒吸(概率很低),机械泵关机后没有进行放气,导致管路内部负压,倒吸泵油,仪器关机,机械泵关机后需要进行放气,拧松放气阀,待听到空气进去的声音结束,拧紧放气阀,放气动作完成,注意B和C型号的仪器(可从仪器电源开关旁边铭牌检查订货号),需要仪器关机半小时以后,才能关闭机械泵电源再进行放气。
测试点数多少取决于待测样品总吸附量以及投气量的大小设置,若想要增加或者减少点数,可以从质量或者投气量改进。需要注意的是点数太少,测试时间会短,但是曲线会不平滑;点数太多,曲线会更平滑,但是测试时间会很长,需要自行调整。
1) 在符合称样量要求的情况下,增加或者减少样品量,会导致点数相应增加或者减少
2) 固定样品量的情况下,可以调整投气量,且能单独调整某个压力区间内的点数多少,增加或者减小投气量,会导致点数相应减少或者增加,注意投气量不得低于1kPa
1) 样品本身孔道复杂,吸附比较慢,导致该压力点测试时间长,常见于微孔材料,属于正常现象
2) 若结构图显示在进气阶段,则代表该气体气源不足,检查恢复即可
3) 存在漏气,可打开吸脱附曲线观察压力变化,若平衡曲线压力呈缓慢上涨趋势,则代表有外界空气进入导致压力升高,多为样品管没有装好或者样品管或者密封圈有破损导致漏气,需要手动停止实验等待实验结束,恢复常压后逐项排查问题原因并解除
1) 检查之前的测试数据是否独立保存在其他盘,若保存在软件安装时默认的保存路径,需要提前备份,以免重装软件时被覆盖,导致测试数据丢失
2) 记录每个工作站对应的Vd,查找方式为打开软件-测试设置-实验参数最上方
3) 确认测试模板都已经备份留存
4) 拍照记录工作站信息,在新建工作站界面打开后,记录名称,订货号,通讯地址
5) 新装软件后,按照上一条记录的信息,重新配置工作站,并确认电脑IP地址修改正确,修改方式可以参考问题14,并确认电脑电源选项为接通电源时不休眠不关机
6) 进入工程模式修改对应工作站的Vd
1) 仪器电源没有打开,检查仪器是否正常供电并打开电源开关
2) 网线没有正常连接,检查网线接口是否松动,重新插拔即可,以及确认网线连接的是当前电脑的主机
3) 电脑IP地址不正确,可以打开软件,在配置工作站界面检查软件的IP地址,2020年后的仪器IP地址默认都是192.168.90.3(若有多台仪器,则最后一位有变化),改电脑IP地址时仅改最后一位即可,确认最后一位大于10,例如192.168.90.90
1) 若在充气阶段出现,则因为气速过快,检查是否气瓶减压表进气压力被误操作调大,一般要求0.08-0.1MPa
2) 若在吸附过程出现,则检查日志,观察是否P₀在缓慢上升,多数为测试时间太长液氮不够导致温度升高,氮气(N₂)脱附出来导致超压
通过结构图或者日志记录检查仪器当前状态,
1) 若卡在进气阶段,检查对应的氮气(N₂)或者氦气是否正确打开减压表及气瓶开关,气瓶是否还有足够的气体
2) 若卡在抽真空阶段,可能原因有机械泵未开启,或者有漏气情况,漏气原因通常为机械泵放气阀未拧紧,样品管未装好拧紧导致漏气,样品管或者密封圈有破损漏气,密封圈和金属垫环上下方向顺序放置错误
预抽开始时,真空泵会有几秒的声音较大及冒白烟情况,属于正常现象,但是若长时间处于这种情况,则代表真空泵一直在抽空气,真空管路有直通空气情况。
检查是否未正确安装样品管或者金属堵头就开始预抽,检查机械泵放气阀是否拧紧,仪器冷阱位置冷阱管是否安装且无破损,检查样品管是否有破损,检查真空泵波纹管是否有裂痕破损,逐项排查回恢复即可
通过结构图或者日志记录检查仪器当前状态,
1) 若结构图处在抽真空阶段,则基本是存在漏气现象,之前测试正常情况下,突然漏气,停止实验后恢复至常压进行检查;多数是样品管没有装好,重装即可,或者样品管和密封圈有破损情况导致漏气,更换新样品管或者密封圈即可
2) 若结构图处在进气阶段,则代表气源不足,检查减压表是否完全打开或者气瓶是否还有气
立即停止预抽,取下加热包停止加热,样品管恢复室温后充气至常压,取下样品管。拆下样品管卡头,清理过滤芯。
抽飞原因为预处理操作不规范,需要规范操作,装好样品管后点击预抽,压力低于0.1kPa后再套上加热包设置加热程序,开始加热;若样品含水量较大,需要提前烘箱105℃烘干1-2小时再放到仪器上进行处理,设置加热程序时要程序升温,在90℃下保温一小时以后再升到目标温度进行处理;若所选型号为非程序升温模式,则需要压力抽到0.1kPa以下以后,再等待半小时后,启动加热;若样品特别轻,测试设置里真空抽速选择“超细粉末”模式。
装样结束后抽出漏斗时速度过快导致,需要缓慢抽出漏斗
1) 液氮杯未上升就提示P₀异常,多为氮气进气速度过快,可能有人调整过减压表出口压力,可以降低氮气进气压力,要求0.08-0.1MPa
2) 液氮杯下降后再次上升,然后提示P₀异常(若是微孔测试手动测Q,实验结束前无下降过程),检查日志记录,P₀显示明显高于常压甚至133kPa,可能原因P₀管未放进液氮杯,液氮量不够,液氮纯度不是高纯,对应检查并解决;还有可能是更换气瓶后,氦气瓶接到氮气进气位置,导致进氮气时实际进的是氦气,更换气瓶后,纯化气路3次以上再进行实验
3) 日志检查P₀明显低于当地大气压,一种可能是氮气不纯常见于实验过程前期,检查氮气纯度或者更换确定是高纯的氮气
1) 样品预处理不充分,测试时有气体挥发出来,需要在不破坏样品的前提下增加处理温度或者延长处理时间
2) 称样量太多或者投气量太小,对应的减小称样量或者增大投气量
3) 先测试Q值(冷自用空间系数)导致氦中毒,去掉自动测试Q值,测试结束后手动后测Q值
一般要求待测面积在5-50m2之间,以10-20最佳,待测面积=比表面积x质量,称量建议如下表:
| 比表面积(m2/g) | <1 | 1-5 | 5-10 | 10-50 | 50-100 | >100 |
|---|---|---|---|---|---|---|
| 质量/g | 10-20g | 10-4g | 4-2g | 2-0.4g | 0.4-0.15g | 0.1g |
C值大于0;线性因子大于0.999,若比表面积比较小,且样品很轻,受限于称样量的情况,线性因子可能只能达到0.995;选点范围内至少有5个点
首先需要明确一个概念,不同原理的仪器,同原理不同配置的仪器,同配置同原理不同厂家的仪器,都可能会存在误差,数据有所区别是正常现象
同型号的仪器对比测试结果有明显区别,原因有多种可能,常见的以下几种:
1) 样品本身均匀性不好,即使同一包样品可能前后两次取出的样品本身就会有区别,测试结果有区别时,可通过交换测试位置重新处理重新测试进行排除,若结果随着位置改变而改变,则确定是样品问题,若结果随着工作站改变,则代表样品本身无问题
2) 样品预处理条件不一致,同样的样品,预处理条件不一样,对结果也有可能会造成较大影响,若需对数,需要确保样品预处理条件完全一致,最好也确保称样量基本一致
3) 检查测试设置是否完全一致,尤其是Q值,P₀,以及报告设置内的选点范围,Vd是否被人误操作改动过
4) 天平误差,人为操作误差,操作称量是否规范,尤其是质量,也会造成结果误差,比表面积越大的样品,质量误差导致的结果差距会越大
使用脉冲式排气,仪器进气卡套接头用扳手松开,手指堵住出气口,然后打开气瓶开关,调整压力到0.4-0.6MPa,关闭分压阀,松开手指,待气体排干净以后,再次堵住,重复开气放气过程,管路3米长,重复5次,管路每延长一米,额外增加2次重复过程,排气完成后分压调至初始要求压力,拧紧卡套接头,纯化气路3次以后再进行实验。
可能原因:1.样品称样量太少;2.预处理不充分,样品未处理干净;3.选点范围不适合;4.微孔样品先测试Q值,导致样品氦中毒
解决方案:1.增大称样量,在不超过体积要求时尽可能多的称量样品;2.在不破坏样品的前提下,增加处理温度,延长处理时间;3.P/P₀推荐选点范围:介孔材料0.05-0.3,微孔材料0.005-0.1,微介孔材料0.01-0.2,可根据实际情况进行左右调整,使得C值和线性满足要求;4.微孔材料样品,测试时选择手动测试Q值,并更新到测试数据中再检查C值和线性
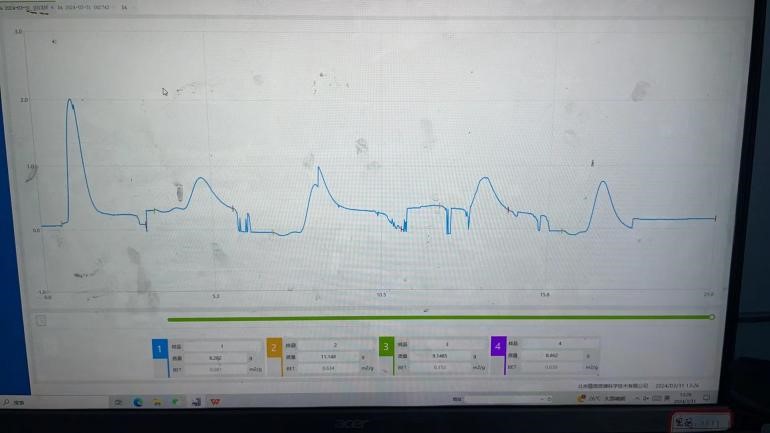
1) 仪器现场地线未接或者不良,波动大
2) 周围有电磁干扰或者大功率设备干扰
若样品结果随着时间变长而慢慢变大,多数因为标样位置的标样长期使用未更换导致,重新称量标样或者此标样用脱气机150℃加热抽真空一小时处理,无脱气机可以倒在干燥皿上,烘箱150℃加热2小时,然后重新确定质量再进行测试
立即关闭仪器电源,关闭软件,重新打开仪器电源及软件,建立连接后电梯自动下降,拿走防挥发盖后重新测试
若关机时样品管已经被顶碎或者电梯被顶变形,按照上述操作重连后,下降结束清理样品管碎渣时注意做好防护,电梯变形后手动稍微弯折回正,可以暂时应急使用,但问题严重的情况,托盘放上液氮杯后可能出现不平的情况,需要更换
此液体基本不可能是水分,水分在真空加热条件下比较容易处理,多为有机物,此情况银粉等生产过程添加有机溶剂的样品容易出现;有其他颜色代表样品也挥发出水分之外的东西
此种情况基本都会对样品本身的结构造成影响,导致测试结果异常,需要降低处理温度,确保不破坏样品结构
1) 进气压力过大,调整减压阀降低进气压力
2) 进气速度过快,调整流量计调低进气速度
1) 确定当地大气压,表压数值的极限真空和当地大气压有关,当地大气压越低,越不容易抽下去
2) 检查真空泵是否启动,开关是否打开,电源线是否连接,是否有管路断开情况
3) 存在漏气,样品管未插紧,油封位置漏气,样品管有裂痕等导致漏气
4) 进气开关没有关闭,导致一边进气一边抽真空



抽飞严重时需要进行清理堆积在油封位置的样品,否则同意导致漏气,真空抽不下去。拆卸顺序按照上图,拆到最后时只剩下油封需要清理,若有新的油封更换,可以用十字螺丝刀将油封翘出,若无新油封更换,可以用气体吹扫,将样品吹干净,再依次装回即可
1) 进入工程模式,拍照记录测试设置,重装软件后对应修改
2) 确认测试数据文件是否在软件目录文件夹下,若在需要备份到其他位置,以免安装时被覆盖导致测试数据丢失
3) 安装新软件后需要重新导入授权文件,导入方式为打开软件,设置-设置-序列号-导入-选中文件,确定即可;或者打开软件-import-选中这个文件,确定即可
4) 打开后,根据型号(可查看电源键旁边的仪器铭牌)建立对应的工作站,修改测试设置内参数后,即可正常测试
预热结束后,检查基线是否走平,确认基线平稳后可以调整调零旋钮调整零点,位置在面向仪器进气口旁边两个旋钮,一个粗调一个微调,可以进行调整基线零点
减少称样量,确保样品在样品管内部均匀分布,称样量最多不能超过样品管管肚的2/3,特别轻的样品不能超过1/2
操作人员温控表误操作上锁,长按set键(最左边的按键),出现菜单界面后点按set键,找到loc选项,将下边的数值调整为0,然后静等几秒自行确认恢复初始界面。
1) 实验室电路没有接地或者接地不良,导致基线波动
2) 电脑主机本身带电,可用电笔检查,若有电,引出接地即可
1) 检查峰型是否正常,峰起始点之前若有明显朝上的凸起,则代表仪器存在漏气,常见为样品管没有装紧或者密封圈位置堆积样品导致漏气
2) 增加样品量,特别轻的粉末按照最多样品管管肚体积的1/2称取,密度大的样品可以按照2/3称取
1) 仪器电源没打开
2) 检查USB线是否松动没接好,重新插拔安装,清理接口位置
3) 电脑USB接口有问题,更换接口位置
4) 重复打开软件
1) 样品管安装时没有竖直安装拧紧,需要竖直向上插入拧紧
2) 拧紧过程中没有两边同时拧紧,先拧紧一边再拧紧另一边导致卡碎,需要两边同时拧紧
3) 装管时用劲过大,掰碎样品管
4) 卡头间距异常,平常拆卸卡头时导致位置偏离,需要联系工程师远程指导重新调整卡头间距
1) SAS软件液氮杯降下来以后,未等实验结束的弹窗出现就直接关机,导致样品管内脱附出来的氮气无法排出,压力升高,把样品管冲下来
2) 切峰版本软件,实验结束后弹窗出现以后需要至少等待5分钟后,才能关机
1) 气体快使用完,导致流量降低,吸附变慢,气体压力低于1MPa后就需要更换新气体
2) 有些样品本身吸附比较慢,导致时间较长,可以减小称样量,装样时保持样品平铺在样品管底部
3) 样品含水量较多,测试前没有处理样品,需要测试前通过专用脱气机进行样品预处理,或者用烘箱烘干
1) 检查是否没有准备液氮或者液氮不够导致无法进行吸附
2) 更换气体后出现,可能是混气更换成了纯气,检查气体标识
3) 测试过程中没有气体通过,检查气瓶阀门是否完全打开,分压表是否调整为适用压力
1) 检查放大倍数是否被改过,默认放大倍数是4
2) 测试漏气导致积分过程把漏气峰计算在内,检查样品管,密封圈是否破损或者未拧紧
3) 如果是标样位置峰面积变小,可能是标样使用时间过长,需要重新加热处理一遍再使用,加热结束使用前需要重新复核质量
4) 电脑配置低,使用久了实验过程有卡顿导致积分异常,清理缓存或者更换电脑
1) 样品管没有装好导致漏气,需要停止实验后重装样品管
2) 仪器开机没有预热就直接测试,需要通气后开机预热至少半小时再进行实验
3) 样品管卡头位置有样品堆积导致漏气,拆下卡头进行清理,如果破损严重需要更换新密封圈
4) 减压表供气不稳定,供气过程中压力表压力波动,需要更换质量好点的减压表
5) 电路波动,外界共用电路的大功率设备启停,或者实验室电路接地不良,根据情况排查即可
1) 没开通气源,开机就显示0,毫无波动,打开气源,并确认调整到指定进气压力
2) 漏气,检查气瓶到仪器之间是否漏气,仪器内部,测试过程中出现此现象,可能是样品管安装不规范或者有破损,密封圈漏气
3) 检查气体比例是否正常,要求氮气(N₂):氦气=3:7
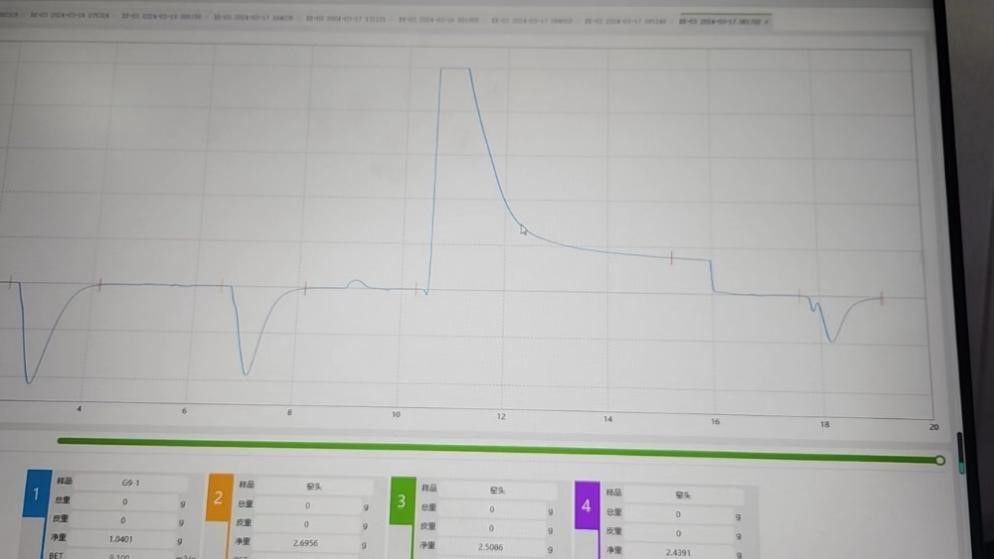
可能原因:测试过程中存在漏气,凸起峰越高,漏气越严重
1)检查样品管卡头密封圈是否变脏,清理干净或者更换密封圈
2)检查样品管是否装好或者有损坏,重装或者更换新的样品管
仪器关机后,第二天或者更久后使用,开机前确保先开通气路,然后开机通电,打开软件仪器预热半小时,检查基线是否走平,走平后可进行实验,未走平需要继续预热。若预热半小时后基线波动很大,则检查仪器和气瓶之间是否漏气。
使用脉冲式排气,仪器进气卡套接头用扳手松开,手指堵住出气口,然后打开气瓶开关,调整压力到0.4-0.6mpa,关闭分压阀,松开手指,待气体排干净以后,再次堵住,重复开气放气过程,管路3米长,重复5次,管路每延长一米,额外增加2次重复过程,排气完成后分压调至初始要求压力,拧紧卡套接头,预热半小时后检查基线是否平稳,平稳后方可进行实验。
服务电话
精微高博公众号
Sales@jwgb.net
 京公网安备 11011202004400号
京公网安备 11011202004400号